

Introduction¶
LaminDB is an open-source data framework for better computational biology. It lets you track data transformations, curate datasets, manage metadata, and query a built-in database for common biological data types.
Why?

Reproducing analytical results or understanding how a dataset or model was created can be a pain. Leave alone training models on historical data, orthogonal assays, or datasets generated by other teams.
Biological datasets are typically managed with versioned storage systems (file systems, object storage, git, dvc), UI-focused community or SaaS platforms, structureless data lakes, rigid data warehouses (SQL, monolithic arrays), and data lakehouses for tabular data.
LaminDB goes beyond these systems with a lakehouse that models biological datasets beyond tables with enough structure to enable queries and enough freedom to keep the pace of R&D high.
For data structures like DataFrame, AnnData, .zarr, .tiledbsoma, etc., LaminDB tracks and provides the rich context that collaborative biological research requires:
data lineage: data sources and transformations; scientists and machine learning models
domain knowledge and experimental metadata: the features and labels derived from domain entities
In this blog post, we discuss a breadth of data management problems of the field.
LaminDB specs
Any LaminDB instance comes with an underlying SQL metadata database to organize files, folders, and arrays across any number of storage locations.
The following detailed specs are for the Python package lamindb. For the analogous R package laminr, see the R docs.
Manage data & metadata with a unified API (“lakehouse”).
Model files and folders as datasets & models via one class:
ArtifactUse array formats in memory & storage: DataFrame, AnnData, MuData, tiledbsoma, … backed by parquet, zarr, tiledb, HDF5, h5ad, DuckDB, …
Create iterable & queryable collections of artifacts with data loaders:
CollectionVersion artifacts, collections & transforms:
IsVersioned
Track data lineage across notebooks, scripts, pipelines & UI.
Track scripts & notebooks with a simple method call:
track()Track functions with a decorator:
tracked()A unified registry for all your notebooks, scripts & pipelines:
TransformA unified registry for all data transformation runs:
RunManage execution reports, source code and Python environments for notebooks & scripts
Integrate with workflow managers: redun, nextflow, snakemake
Manage registries for experimental metadata & in-house ontologies, import public ontologies.
Use >20 public ontologies with module
bionty:Gene,Protein,CellMarker,ExperimentalFactor,CellType,CellLine,Tissue, …Use a canonical wetlab database schema module
wetlabSafeguards against typos & duplications
Version ontology
Validate, standardize & annotate.
Validate & standardize metadata:
validate,standardize.High-level curation flow including annotation:
CuratorInspect validation failures:
inspect
Organize and share data across a mesh of LaminDB instances.
Create & connect to instances with the same ease as git repos:
lamin init&lamin connectZero-copy transfer data across instances
Integrate with analytics tools.
Vitessce:
save_vitessce_config
Zero lock-in, scalable, auditable.
Zero lock-in: LaminDB runs on generic backends server-side and is not a client for “Lamin Cloud”
Flexible storage backends (local, S3, GCP, https, HF, R2, anything fsspec supports)
Two SQL backends for managing metadata: SQLite & Postgres
Scalable: metadata registries support 100s of millions of entries, storage is as scalable as S3
Plug-in custom schema modules & manage database schema migrations
Auditable: data & metadata records are hashed, timestamped, and attributed to users (full audit log to come)
Secure: embedded in your infrastructure
Tested, typed, idempotent & ACID
LaminHub is a data collaboration hub built on LaminDB similar to how GitHub is built on git.
LaminHub overview
See for yourself by browsing the demo instances in the hub UI or lamin connect owner/instance them via the CLI.
lamin.ai/laminlabs/lamindata - A generic demo instance with various data types
lamin.ai/laminlabs/cellxgene - An instance that interfaces the CELLxGENE data (guide)
lamin.ai/laminlabs/arrayloader-benchmarks - Work with ML models & benchmarks
See the pricing page. Basic LaminHub features are free.
Secure & intuitive access management.
Rather than configuring storage & database permissions directly on AWS or GCP, LaminHub allows you to manage collaborators for databases & storage locations in the same way you manage access to repositories on GitHub. See Access management.
A UI to work with LaminDB instances.
See an overview of all datasets, models, code, and metadata in your instance.
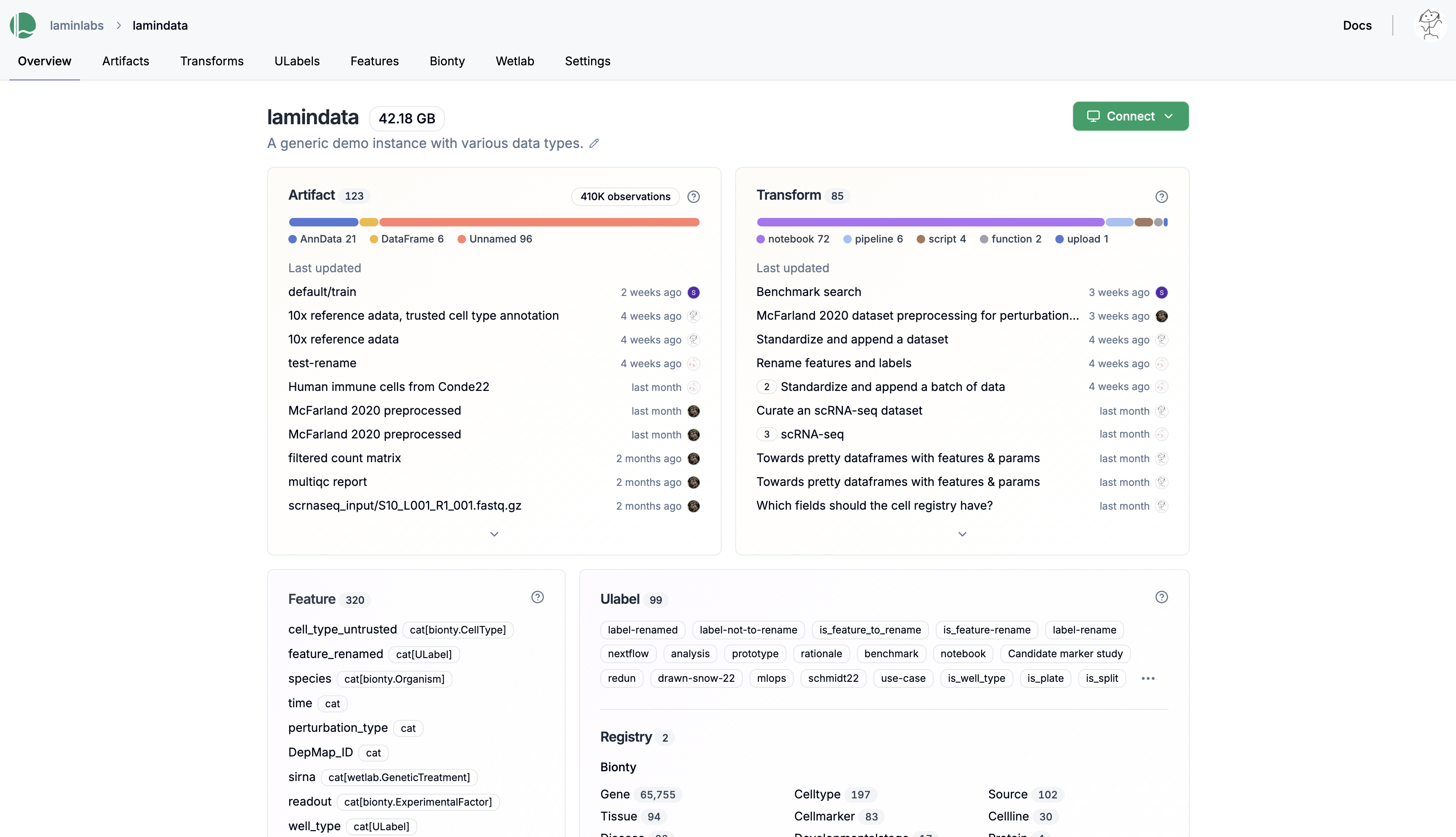
See validated datasets in context of ontologies & experimental metadata.
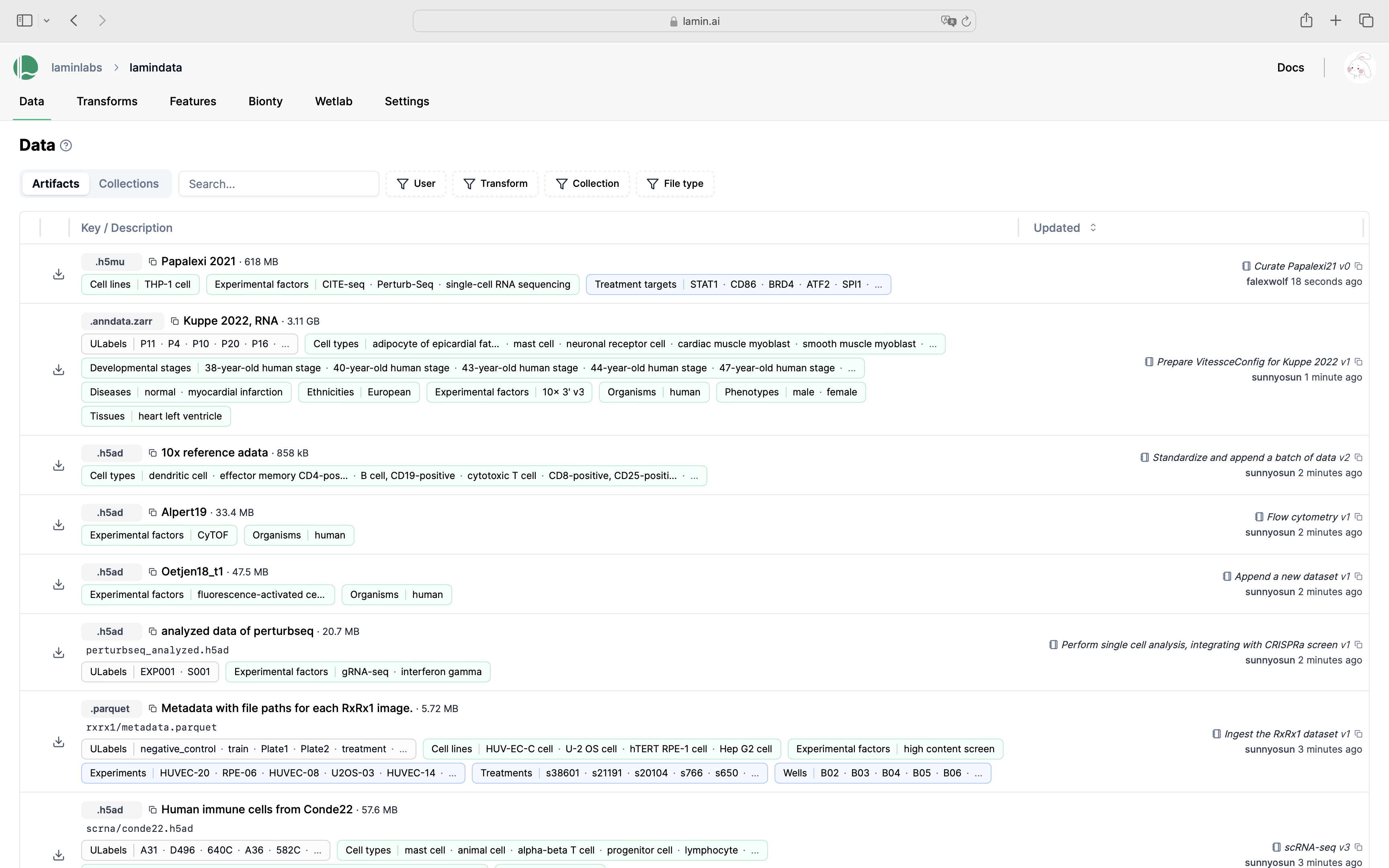
Query & search.
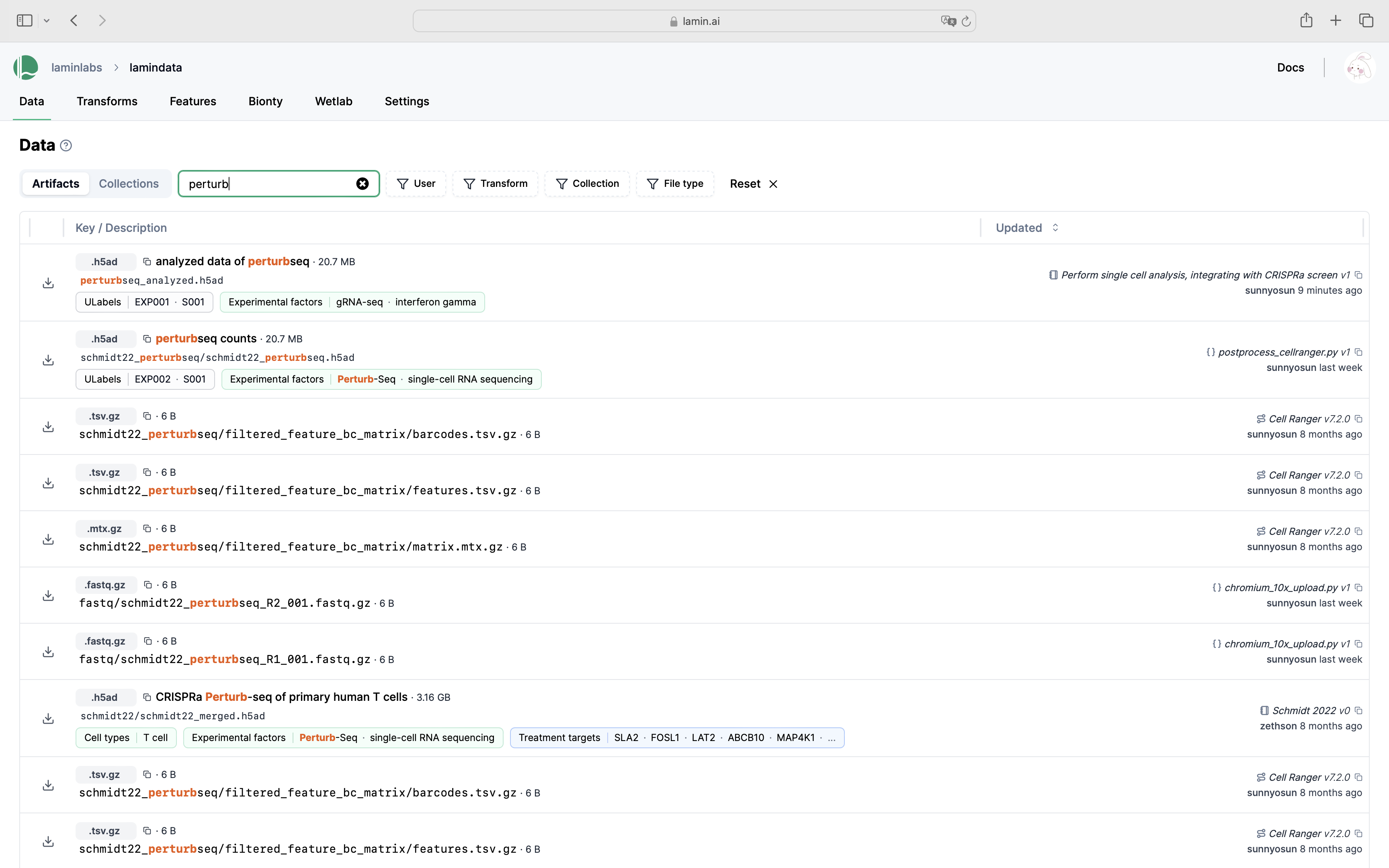
See scripts, notebooks & pipelines with their inputs & outputs.

Track pipelines, notebooks & UI transforms in one place.
Quickstart¶
Install the lamindb Python package.
pip install 'lamindb[jupyter,bionty]' # support notebooks & biological ontologies
Connect to a LaminDB instance.
lamin connect account/instance # <-- replace with your instance
Access an input dataset and save an output dataset.
import lamindb as ln
ln.track() # track a run of your notebook or script
artifact = ln.Artifact.get("3TNCsZZcnIBv2WGb0001") # get an artifact by uid
filepath = artifact.cache() # cache the artifact on disk
# do your work
ln.Artifact("./my_dataset.csv", key="my_results/my_dataset.csv").save() # save a file
ln.finish() # mark the run as finished & save a report for the current notebook/script
# laminr needs pip install 'lamindb'
install.packages("laminr", dependencies = TRUE) # install the laminr package from CRAN
library(laminr)
db <- connect() # connect to the instance you configured on the terminal
db$track() # track a run of your notebook or script
artifact <- db$Artifact$get("3TNCsZZcnIBv2WGb0001") # get an artifact by uid
filepath <- artifact$cache() # cache the artifact on disk
# do your work
db$Artifact.from_path("./my_dataset.csv", key="my_results/my_dataset.csv").save() # save a folder
db$finish() # mark the run finished
Depending on whether you ran RStudio’s notebook mode, you may need to save an html export for .qmd or .Rmd file via the command-line.
lamin save my-analysis.Rmd
For more, see the R docs.
Walkthrough¶
A LaminDB instance is a database that manages metadata for datasets in different storage locations. Let’s create one.
!lamin init --storage ./lamin-intro --modules bionty
Show code cell output
! using anonymous user (to identify, call: lamin login)
→ initialized lamindb: anonymous/lamin-intro
You can also pass a cloud storage location to --storage (S3, GCP, R2, HF, etc.) or a Postgres database connection string to --db. See Install & setup.
If you decide to connect your LaminDB instance to the hub, you will see data & metadata in a UI.
On the hub.
Transforms¶
A data transformation (a “transform”) is a piece of code (script, notebook, pipeline, function) that can be applied to input data to produce output data.
When you call track() in a script or notebook, inputs, outputs, source code, run reports and environments start to get automatically tracked.
import lamindb as ln
import pandas as pd
ln.track() # track the current notebook or script
Show code cell output
→ connected lamindb: anonymous/lamin-intro
→ created Transform('SYMwNSmV0XgW0000'), started new Run('6ip2Vjpz...') at 2025-02-26 20:55:24 UTC
→ notebook imports: anndata==0.11.3 bionty==1.1.0 lamindb==1.1.0 pandas==2.2.3
Is this compliant with OpenLineage?
Yes. What OpenLineage calls a “job”, LaminDB calls a “transform”. What OpenLineage calls a “run”, LaminDB calls a “run”.
You can see all your transforms and their runs in the Transform and Run registries.
ln.Transform.df()
Show code cell output
| uid | key | description | type | source_code | hash | reference | reference_type | space_id | _template_id | version | is_latest | created_at | created_by_id | _aux | _branch_code | |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| id | ||||||||||||||||
| 1 | SYMwNSmV0XgW0000 | introduction.ipynb | Introduction | notebook | None | None | None | None | 1 | None | None | True | 2025-02-26 20:55:24.290000+00:00 | 1 | None | 1 |
ln.Run.df()
Show code cell output
| uid | name | started_at | finished_at | reference | reference_type | _is_consecutive | _status_code | space_id | transform_id | report_id | _logfile_id | environment_id | initiated_by_run_id | created_at | created_by_id | _aux | _branch_code | |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| id | ||||||||||||||||||
| 1 | 6ip2VjpzqFx6aRFrlYhD | None | 2025-02-26 20:55:24.303762+00:00 | None | None | None | None | 0 | 1 | 1 | None | None | None | None | 2025-02-26 20:55:24.304000+00:00 | 1 | None | 1 |
Artifacts¶
An Artifact stores a dataset or model as a file or folder.
# an example dataset
df = ln.core.datasets.small_dataset1(otype="DataFrame", with_typo=True)
df
Show code cell output
| ENSG00000153563 | ENSG00000010610 | ENSG00000170458 | cell_medium | sample_note | cell_type_by_expert | cell_type_by_model | |
|---|---|---|---|---|---|---|---|
| sample1 | 1 | 3 | 5 | DMSO | was ok | B cell | B cell |
| sample2 | 2 | 4 | 6 | IFNJ | looks naah | T cell | T cell |
| sample3 | 3 | 5 | 7 | DMSO | pretty! 🤩 | T cell | T cell |
# create & save an artifact from a DataFrame
artifact = ln.Artifact.from_df(df, key="my_datasets/rnaseq1.parquet").save()
# describe the artifact
artifact.describe()
Show code cell output
Artifact .parquet/DataFrame └── General ├── .uid = 'kXxKsHYfz32dOrSY0000' ├── .key = 'my_datasets/rnaseq1.parquet' ├── .size = 6586 ├── .hash = 'TZbCzj4ZBX_KxhpwNXq25g' ├── .n_observations = 3 ├── .path = /home/runner/work/lamin-docs/lamin-docs/docs/lamin-intro/.lamindb/kXxKsHYfz32dOrSY0000.parquet ├── .created_by = anonymous ├── .created_at = 2025-02-26 20:55:24 └── .transform = 'Introduction'
Copy or download the artifact into a local cache.
artifact.cache()
Show code cell output
PosixUPath('/home/runner/work/lamin-docs/lamin-docs/docs/lamin-intro/.lamindb/kXxKsHYfz32dOrSY0000.parquet')
Open the artifact for streaming.
dataset = artifact.open() # returns pyarrow.Dataset
dataset.head(2).to_pandas()
Show code cell output
| ENSG00000153563 | ENSG00000010610 | ENSG00000170458 | cell_medium | sample_note | cell_type_by_expert | cell_type_by_model | |
|---|---|---|---|---|---|---|---|
| sample1 | 1 | 3 | 5 | DMSO | was ok | B cell | B cell |
| sample2 | 2 | 4 | 6 | IFNJ | looks naah | T cell | T cell |
Cache & load the artifact into memory.
artifact.load()
Show code cell output
| ENSG00000153563 | ENSG00000010610 | ENSG00000170458 | cell_medium | sample_note | cell_type_by_expert | cell_type_by_model | |
|---|---|---|---|---|---|---|---|
| sample1 | 1 | 3 | 5 | DMSO | was ok | B cell | B cell |
| sample2 | 2 | 4 | 6 | IFNJ | looks naah | T cell | T cell |
| sample3 | 3 | 5 | 7 | DMSO | pretty! 🤩 | T cell | T cell |
View data lineage.
artifact.view_lineage()

How do I create an artifact for a file or folder?
Source path is local:
ln.Artifact("./my_data.fcs", key="my_data.fcs")
ln.Artifact("./my_images/", key="my_images")
Upon artifact.save(), the source path will be copied or uploaded into your instance’s current storage, visible & changeable via ln.settings.storage.
If the source path is remote or already in a registered storage location, artifact.save() won’t trigger a copy or upload but register the existing path.
ln.Artifact("s3://my-bucket/my_data.fcs") # key is auto-populated from S3, you can optionally pass a description
ln.Artifact("s3://my-bucket/my_images/") # key is auto-populated from S3, you can optionally pass a description
You can also use other remote file systems supported by `fsspec`.
How does LaminDB compare to a AWS S3?
LaminDB provides a database on top of AWS S3 (or GCP storage, file systems, etc.).
Similar to organizing files with paths, you can organize artifacts using the key parameter of Artifact.
However, you’ll see that you can more conveniently query data by entities you care about: people, code, experiments, genes, proteins, cell types, etc.
Are artifacts aware of array-like data?
Yes.
You can make artifacts from paths referencing array-like objects:
ln.Artifact("./my_anndata.h5ad", key="my_anndata.h5ad")
ln.Artifact("./my_zarr_array/", key="my_zarr_array")
Or from in-memory objects:
ln.Artifact.from_df(df, key="my_dataframe.parquet")
ln.Artifact.from_anndata(adata, key="my_anndata.h5ad")
You can open large artifacts for slicing from the cloud or load small artifacts directly into memory.
Just like transforms, artifacts are versioned. Let’s create a new version by revising the dataset.
# keep the dataframe with a typo around - we'll need it later
df_typo = df.copy()
# fix the "IFNJ" typo
df["cell_medium"] = df["cell_medium"].cat.rename_categories({"IFNJ": "IFNG"})
# create a new version
artifact = ln.Artifact.from_df(df, key="my_datasets/rnaseq1.parquet").save()
# see all versions of an artifact
artifact.versions.df()
Show code cell output
→ creating new artifact version for key='my_datasets/rnaseq1.parquet' (storage: '/home/runner/work/lamin-docs/lamin-docs/docs/lamin-intro')
| uid | key | description | suffix | kind | otype | size | hash | n_files | n_observations | _hash_type | _key_is_virtual | _overwrite_versions | space_id | storage_id | schema_id | version | is_latest | run_id | created_at | created_by_id | _aux | _branch_code | |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| id | |||||||||||||||||||||||
| 1 | kXxKsHYfz32dOrSY0000 | my_datasets/rnaseq1.parquet | None | .parquet | dataset | DataFrame | 6586 | TZbCzj4ZBX_KxhpwNXq25g | None | 3 | md5 | True | False | 1 | 1 | None | None | False | 1 | 2025-02-26 20:55:24.826000+00:00 | 1 | None | 1 |
| 2 | kXxKsHYfz32dOrSY0001 | my_datasets/rnaseq1.parquet | None | .parquet | dataset | DataFrame | 6586 | 6iyId6A4cyhKxhls4UxsQA | None | 3 | md5 | True | False | 1 | 1 | None | None | True | 1 | 2025-02-26 20:55:25.001000+00:00 | 1 | None | 1 |
Can I also create new versions independent of key?
That works, too, you can use revises:
artifact_v1 = ln.Artifact.from_df(df, description="Just a description").save()
# below revises artifact_v1
artifact_v2 = ln.Artifact.from_df(df_updated, revises=artifact_v1).save()
The good thing about passing revises: Artifact is that you don’t need to worry about coming up with naming conventions for paths.
The good thing about versioning based on key is that it’s how all data versioning tools are doing it.
Labels¶
Annotate an artifact with a ULabel and a bionty.CellType. The same works for any entity in any custom schema module.
import bionty as bt
# create & save a typed label
experiment_type = ln.ULabel(name="Experiment", is_type=True).save()
candidate_marker_experiment = ln.ULabel(
name="Candidate marker experiment", type=experiment_type
).save()
# label the artifact
artifact.ulabels.add(candidate_marker_experiment)
# repeat for a bionty entity
cell_type = bt.CellType.from_source(name="effector T cell").save()
artifact.cell_types.add(cell_type)
# describe the artifact
artifact.describe()
Show code cell output
Artifact .parquet/DataFrame ├── General │ ├── .uid = 'kXxKsHYfz32dOrSY0001' │ ├── .key = 'my_datasets/rnaseq1.parquet' │ ├── .size = 6586 │ ├── .hash = '6iyId6A4cyhKxhls4UxsQA' │ ├── .n_observations = 3 │ ├── .path = /home/runner/work/lamin-docs/lamin-docs/docs/lamin-intro/.lamindb/kXxKsHYfz32dOrSY0001.parquet │ ├── .created_by = anonymous │ ├── .created_at = 2025-02-26 20:55:25 │ └── .transform = 'Introduction' └── Labels └── .cell_types bionty.CellType effector T cell .ulabels ULabel Candidate marker experiment
For annotating datasets with parsed labels like the cell_mediums DMSO & IFNG, jump to “Curate datasets”.
Registries¶
LaminDB’s central classes are registries that store records (Record objects).
The easiest way to see the latest records for a registry is to call the class method df.
ln.ULabel.df()
Show code cell output
| uid | name | is_type | description | reference | reference_type | space_id | type_id | run_id | created_at | created_by_id | _aux | _branch_code | |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| id | |||||||||||||
| 2 | 2R4gTvsM | Candidate marker experiment | False | None | None | None | 1 | 1.0 | 1 | 2025-02-26 20:55:25.208000+00:00 | 1 | None | 1 |
| 1 | zFFbAPVo | Experiment | True | None | None | None | 1 | NaN | 1 | 2025-02-26 20:55:25.203000+00:00 | 1 | None | 1 |
A record and its registry share the same fields, which define the metadata you can query for. If you want to see them, look at the class or auto-complete.
ln.Artifact
Show code cell output
Artifact
Simple fields
.uid: CharField
.key: CharField
.description: CharField
.suffix: CharField
.kind: CharField
.otype: CharField
.size: BigIntegerField
.hash: CharField
.n_files: BigIntegerField
.n_observations: BigIntegerField
.version: CharField
.is_latest: BooleanField
.created_at: DateTimeField
.updated_at: DateTimeField
Relational fields
.space: Space
.storage: Storage
.run: Run
.schema: Schema
.created_by: User
.ulabels: ULabel
.input_of_runs: Run
.feature_sets: Schema
.collections: Collection
.projects: Project
.references: Reference
Bionty fields
.organisms: bionty.Organism
.genes: bionty.Gene
.proteins: bionty.Protein
.cell_markers: bionty.CellMarker
.tissues: bionty.Tissue
.cell_types: bionty.CellType
.diseases: bionty.Disease
.cell_lines: bionty.CellLine
.phenotypes: bionty.Phenotype
.pathways: bionty.Pathway
.experimental_factors: bionty.ExperimentalFactor
.developmental_stages: bionty.DevelopmentalStage
.ethnicities: bionty.Ethnicity
Query & search¶
You can write arbitrary relational queries using the class methods get and filter.
The syntax for it is Django’s query syntax.
# get a single record (here the current notebook)
transform = ln.Transform.get(key="introduction.ipynb")
# get a set of records by filtering on description
ln.Artifact.filter(key__startswith="my_datasets/").df()
# query all artifacts ingested from a transform
artifacts = ln.Artifact.filter(transform=transform).all()
# query all artifacts ingested from a notebook with "intro" in the title and labeled "Candidate marker experiment"
artifacts = ln.Artifact.filter(
transform__description__icontains="intro", ulabels=candidate_marker_experiment
).all()
The class methods search and lookup help with approximate matches.
# search in a registry
ln.Transform.search("intro").df()
# look up records with auto-complete
ulabels = ln.ULabel.lookup()
cell_types = bt.CellType.lookup()
Show me a screenshot
Features¶
You can annotate datasets by associated features.
# define the "temperature" & "experiment" features
ln.Feature(name="temperature", dtype=float).save()
ln.Feature(
name="experiment", dtype=ln.ULabel
).save() # categorical values are validated against the ULabel registry
# annotate
artifact.features.add_values(
{"temperature": 21.6, "experiment": "Candidate marker experiment"}
)
# describe the artifact
artifact.describe()
Show code cell output
Artifact .parquet/DataFrame ├── General │ ├── .uid = 'kXxKsHYfz32dOrSY0001' │ ├── .key = 'my_datasets/rnaseq1.parquet' │ ├── .size = 6586 │ ├── .hash = '6iyId6A4cyhKxhls4UxsQA' │ ├── .n_observations = 3 │ ├── .path = /home/runner/work/lamin-docs/lamin-docs/docs/lamin-intro/.lamindb/kXxKsHYfz32dOrSY0001.parquet │ ├── .created_by = anonymous │ ├── .created_at = 2025-02-26 20:55:25 │ └── .transform = 'Introduction' ├── Linked features │ └── experiment cat[ULabel] Candidate marker experiment │ temperature float 21.6 └── Labels └── .cell_types bionty.CellType effector T cell .ulabels ULabel Candidate marker experiment
Query artifacts by features.
ln.Artifact.features.filter(experiment__contains="marker experiment").df()
Show code cell output
| uid | key | description | suffix | kind | otype | size | hash | n_files | n_observations | _hash_type | _key_is_virtual | _overwrite_versions | space_id | storage_id | schema_id | version | is_latest | run_id | created_at | created_by_id | _aux | _branch_code | |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| id | |||||||||||||||||||||||
| 2 | kXxKsHYfz32dOrSY0001 | my_datasets/rnaseq1.parquet | None | .parquet | dataset | DataFrame | 6586 | 6iyId6A4cyhKxhls4UxsQA | None | 3 | md5 | True | False | 1 | 1 | None | None | True | 1 | 2025-02-26 20:55:25.001000+00:00 | 1 | None | 1 |
The easiest way to validate & annotate a dataset by the features they measure is via a Curator: jump to “Curate datasets”.
Key use cases¶
Understand data lineage¶
Understand where a dataset comes from and what it’s used for (background).
artifact.view_lineage()

I just want to see the transforms.
transform.view_lineage()

You don’t need a workflow manager to track data lineage (if you want to use one, see Pipelines – workflow managers). All you need is:
import lamindb as ln
ln.track() # track your run, start tracking inputs & outputs
# your code
ln.finish() # mark run as finished, save execution report, source code & environment
On the hub.
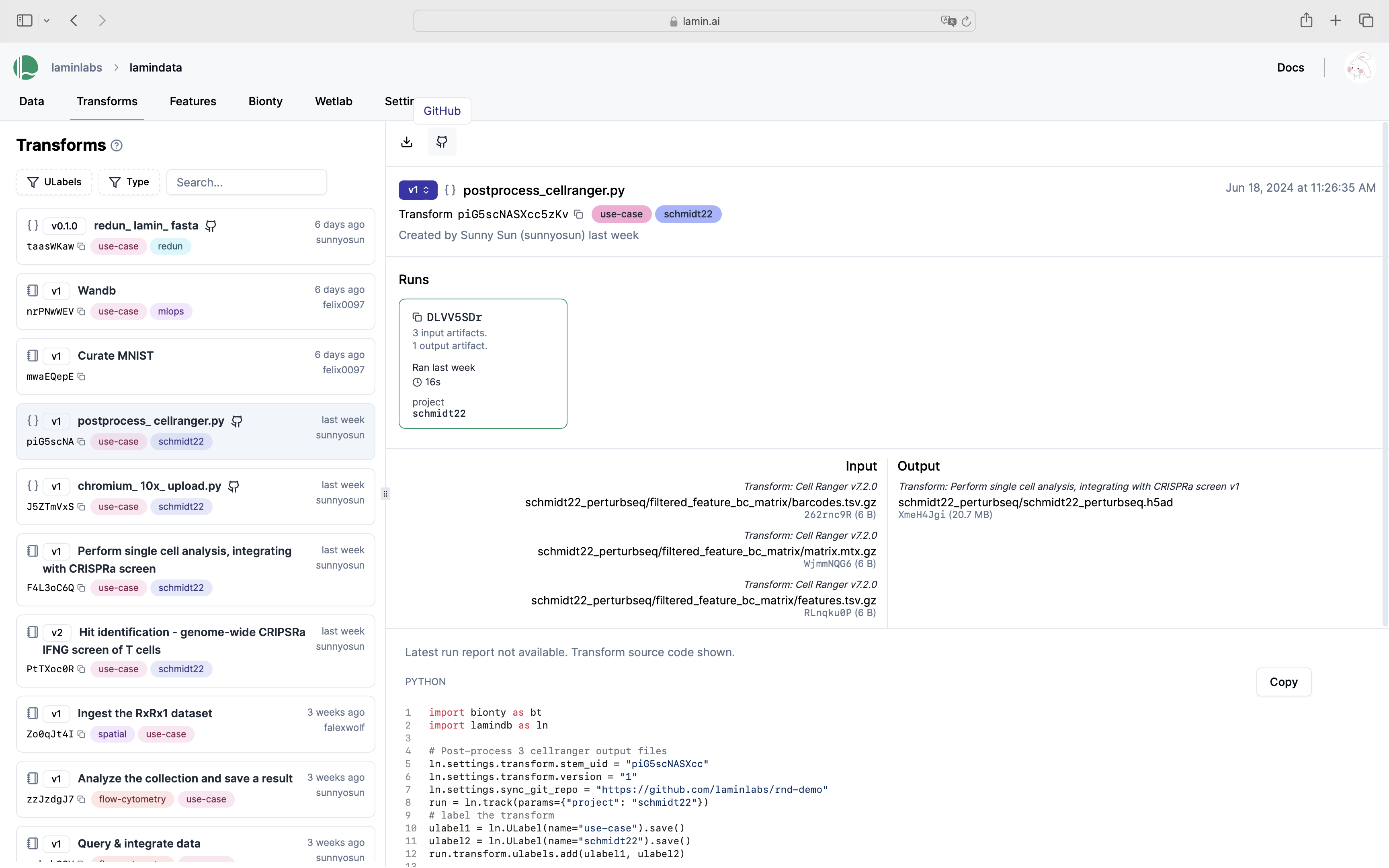
Below is how a single transform (a notebook) with its run report looks on the hub.
To create a new version of a notebook or script, run lamin load on the terminal, e.g.,
$ lamin load https://lamin.ai/laminlabs/lamindata/transform/13VINnFk89PE0004
→ notebook is here: mcfarland_2020_preparation.ipynb
Curate datasets¶
You already saw how to ingest datasets without validation. This is often enough if you’re prototyping or working with one-off studies. But if you want to create a big body of standardized data, you have to invest the time to curate your datasets.
Let’s define a Schema to curate a DataFrame.
# define valid labels
cell_medium_type = ln.ULabel(name="CellMedium", is_type=True).save()
ln.ULabel(name="DMSO", type=cell_medium_type).save()
ln.ULabel(name="IFNG", type=cell_medium_type).save()
# define the schema
schema = ln.Schema(
name="My DataFrame schema",
features=[
ln.Feature(name="ENSG00000153563", dtype=int).save(),
ln.Feature(name="ENSG00000010610", dtype=int).save(),
ln.Feature(name="ENSG00000170458", dtype=int).save(),
ln.Feature(name="cell_medium", dtype=ln.ULabel).save(),
],
).save()
With a Curator, we can save an annotated & validated artifact with a single line of code.
curator = ln.curators.DataFrameCurator(df, schema)
# save curated artifact
artifact = curator.save_artifact(key="my_curated_dataset.parquet") # calls .validate()
# see the parsed annotations
artifact.describe()
# query for a ulabel that was parsed from the dataset
ln.Artifact.get(ulabels__name="IFNG")
Show code cell output
✓ "cell_medium" is validated against ULabel.name
→ found artifact with same hash: Artifact(uid='kXxKsHYfz32dOrSY0001', is_latest=True, key='my_datasets/rnaseq1.parquet', suffix='.parquet', kind='dataset', otype='DataFrame', size=6586, hash='6iyId6A4cyhKxhls4UxsQA', n_observations=3, space_id=1, storage_id=1, run_id=1, created_by_id=1, created_at=2025-02-26 20:55:25 UTC); to track this artifact as an input, use: ln.Artifact.get()
! key my_datasets/rnaseq1.parquet on existing artifact differs from passed key my_curated_dataset.parquet
✓ 4 unique terms (57.10%) are validated for name
! 3 unique terms (42.90%) are not validated for name: 'sample_note', 'cell_type_by_expert', 'cell_type_by_model'
✓ loaded 4 Feature records matching name: 'ENSG00000153563', 'ENSG00000010610', 'ENSG00000170458', 'cell_medium'
! did not create Feature records for 3 non-validated names: 'cell_type_by_expert', 'cell_type_by_model', 'sample_note'
→ returning existing schema with same hash: Schema(uid='G88zs07a70LW6BPSy5v3', name='My DataFrame schema', n=4, itype='Feature', is_type=False, hash='nGK1MnGU-G3l_BfyBJ_n0g', minimal_set=True, ordered_set=False, maximal_set=False, created_by_id=1, run_id=1, space_id=1, created_at=2025-02-26 20:55:26 UTC)
! updated otype from None to DataFrame
Artifact .parquet/DataFrame ├── General │ ├── .uid = 'kXxKsHYfz32dOrSY0001' │ ├── .key = 'my_datasets/rnaseq1.parquet' │ ├── .size = 6586 │ ├── .hash = '6iyId6A4cyhKxhls4UxsQA' │ ├── .n_observations = 3 │ ├── .path = /home/runner/work/lamin-docs/lamin-docs/docs/lamin-intro/.lamindb/kXxKsHYfz32dOrSY0001.parquet │ ├── .created_by = anonymous │ ├── .created_at = 2025-02-26 20:55:25 │ └── .transform = 'Introduction' ├── Dataset features/schema │ └── columns • 4 [Feature] │ cell_medium cat[ULabel] DMSO, IFNG │ ENSG00000153563 int │ ENSG00000010610 int │ ENSG00000170458 int ├── Linked features │ └── experiment cat[ULabel] Candidate marker experiment │ temperature float 21.6 └── Labels └── .cell_types bionty.CellType effector T cell .ulabels ULabel Candidate marker experiment, DMSO, IFNG
Artifact(uid='kXxKsHYfz32dOrSY0001', is_latest=True, key='my_datasets/rnaseq1.parquet', suffix='.parquet', kind='dataset', otype='DataFrame', size=6586, hash='6iyId6A4cyhKxhls4UxsQA', n_observations=3, space_id=1, storage_id=1, run_id=1, schema_id=1, created_by_id=1, created_at=2025-02-26 20:55:25 UTC)
If we feed a dataset with an invalid dtype or typo, we’ll get a ValidationError.
curator = ln.curators.DataFrameCurator(df_typo, schema)
# validate the dataset
try:
curator.validate()
except ln.errors.ValidationError as error:
print(str(error))
Show code cell output
• mapping "cell_medium" on ULabel.name
! 1 term is not validated: 'IFNJ'
→ fix typos, remove non-existent values, or save terms via .add_new_from("cell_medium")
1 term is not validated: 'IFNJ'
→ fix typos, remove non-existent values, or save terms via .add_new_from("cell_medium")
Manage biological registries¶
The generic Feature and ULabel registries will get you pretty far.
But let’s now look at what you do can with a dedicated biological registry like Gene.
Every bionty registry is based on configurable public ontologies (>20 of them).
cell_types = bt.CellType.public()
cell_types
Show code cell output
PublicOntology
Entity: CellType
Organism: all
Source: cl, 2024-08-16
#terms: 2959
cell_types.search("gamma-delta T cell").head(2)
Show code cell output
| name | definition | synonyms | parents | |
|---|---|---|---|---|
| ontology_id | ||||
| CL:0000798 | gamma-delta T cell | A T Cell That Expresses A Gamma-Delta T Cell R... | gamma-delta T-cell|gamma-delta T lymphocyte|ga... | [CL:0000084] |
| CL:4033072 | cycling gamma-delta T cell | A(N) Gamma-Delta T Cell That Is Cycling. | proliferating gamma-delta T cell | [CL:4033069, CL:0000798] |
Define an AnnData schema.
# define var schema
var_schema = ln.Schema(
name="my_var_schema",
itype=bt.Gene.ensembl_gene_id,
dtype=int,
).save()
obs_schema = ln.Schema(
name="my_obs_schema",
features=[
ln.Feature(name="cell_medium", dtype=ln.ULabel).save(),
],
).save()
# define composite schema
anndata_schema = ln.Schema(
name="my_anndata_schema",
otype="AnnData",
components={"obs": obs_schema, "var": var_schema},
).save()
→ returning existing Feature record with same name: 'cell_medium'
Validate & annotate an AnnData.
import anndata as ad
import bionty as bt
# store the dataset as an AnnData object to distinguish data from metadata
adata = ad.AnnData(
df[["ENSG00000153563", "ENSG00000010610", "ENSG00000170458"]],
obs=df[["cell_medium"]],
)
# save curated artifact
curator = ln.curators.AnnDataCurator(adata, anndata_schema)
artifact = curator.save_artifact(description="my RNA-seq")
artifact.describe()
Show code cell output
✓ "cell_medium" is validated against ULabel.name
✓ created 1 Organism record from Bionty matching name: 'human'
• path content will be copied to default storage upon `save()` with key `None` ('.lamindb/g5mV0h4kfKxHIsE40000.h5ad')
✓ storing artifact 'g5mV0h4kfKxHIsE40000' at '/home/runner/work/lamin-docs/lamin-docs/docs/lamin-intro/.lamindb/g5mV0h4kfKxHIsE40000.h5ad'
• parsing feature names of X stored in slot 'var'
✓ 3 unique terms (100.00%) are validated for ensembl_gene_id
✓ linked: Schema(uid='1QdQFbPCntdGOzJGhtDw', n=3, dtype='int', itype='bionty.Gene', is_type=False, hash='f2UVeHefaZxXFjmUwo9Ozw', minimal_set=True, ordered_set=False, maximal_set=False, created_by_id=1, run_id=1, space_id=1, created_at=<django.db.models.expressions.DatabaseDefault object at 0x7f22a1ad7a10>)
• parsing feature names of slot 'obs'
✓ 1 unique term (100.00%) is validated for name
→ returning existing schema with same hash: Schema(uid='wXSgcgUlZx76Nz1Snb1O', name='my_obs_schema', n=1, itype='Feature', is_type=False, hash='3POoh4DkKPQbuWp9wKDkvw', minimal_set=True, ordered_set=False, maximal_set=False, created_by_id=1, run_id=1, space_id=1, created_at=2025-02-26 20:55:27 UTC)
! updated otype from None to DataFrame
✓ linked: Schema(uid='wXSgcgUlZx76Nz1Snb1O', name='my_obs_schema', n=1, itype='Feature', is_type=False, otype='DataFrame', hash='3POoh4DkKPQbuWp9wKDkvw', minimal_set=True, ordered_set=False, maximal_set=False, created_by_id=1, run_id=1, space_id=1, created_at=2025-02-26 20:55:27 UTC)
✓ saved 1 feature set for slot: 'var'
Artifact .h5ad/AnnData ├── General │ ├── .uid = 'g5mV0h4kfKxHIsE40000' │ ├── .size = 19240 │ ├── .hash = '9KxC2jVWsUrREWhcS15_Sg' │ ├── .n_observations = 3 │ ├── .path = /home/runner/work/lamin-docs/lamin-docs/docs/lamin-intro/.lamindb/g5mV0h4kfKxHIsE40000.h5ad │ ├── .created_by = anonymous │ ├── .created_at = 2025-02-26 20:55:29 │ └── .transform = 'Introduction' ├── Dataset features/schema │ ├── var • 3 [bionty.Gene] │ │ CD8A int │ │ CD4 int │ │ CD14 int │ └── obs • 1 [Feature] │ cell_medium cat[ULabel] DMSO, IFNG └── Labels └── .ulabels ULabel DMSO, IFNG
Query for typed features.
# get a lookup object for human genes
genes = bt.Gene.filter(organism__name="human").lookup()
# query for all feature sets that contain CD8A
feature_sets = ln.FeatureSet.filter(genes=genes.cd8a).all()
# write the query
ln.Artifact.filter(feature_sets__in=feature_sets).df()
Show code cell output
| uid | key | description | suffix | kind | otype | size | hash | n_files | n_observations | _hash_type | _key_is_virtual | _overwrite_versions | space_id | storage_id | schema_id | version | is_latest | run_id | created_at | created_by_id | _aux | _branch_code | |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| id | |||||||||||||||||||||||
| 3 | g5mV0h4kfKxHIsE40000 | None | my RNA-seq | .h5ad | dataset | AnnData | 19240 | 9KxC2jVWsUrREWhcS15_Sg | None | 3 | md5 | True | False | 1 | 1 | 4 | None | True | 1 | 2025-02-26 20:55:29.315000+00:00 | 1 | None | 1 |
Update ontologies, e.g., create a cell type record and add a new cell state.
# create an ontology-coupled cell type record and save it
neuron = bt.CellType.from_source(name="neuron").save()
# create a record to track a new cell state
new_cell_state = bt.CellType(
name="my neuron cell state", description="explains X"
).save()
# express that it's a neuron state
new_cell_state.parents.add(neuron)
# view ontological hierarchy
new_cell_state.view_parents(distance=2)
Show code cell output
✓ created 1 CellType record from Bionty matching name: 'neuron'
✓ created 3 CellType records from Bionty matching ontology_id: 'CL:0002319', 'CL:0000404', 'CL:0000393'

Scale learning¶
How do you integrate new datasets with your existing datasets? Leverage Collection.
# a new dataset
df2 = ln.core.datasets.small_dataset2(otype="DataFrame")
adata = ad.AnnData(
df2[["ENSG00000153563", "ENSG00000010610", "ENSG00000004468"]],
obs=df2[["cell_medium"]],
)
curator = ln.curators.AnnDataCurator(adata, anndata_schema)
artifact2 = curator.save_artifact(key="my_datasets/my_rnaseq2.h5ad")
Show code cell output
✓ "cell_medium" is validated against ULabel.name
• path content will be copied to default storage upon `save()` with key 'my_datasets/my_rnaseq2.h5ad'
✓ storing artifact 'xToOIN8U3fzMuQuZ0000' at '/home/runner/work/lamin-docs/lamin-docs/docs/lamin-intro/.lamindb/xToOIN8U3fzMuQuZ0000.h5ad'
• parsing feature names of X stored in slot 'var'
✓ 3 unique terms (100.00%) are validated for ensembl_gene_id
✓ linked: Schema(uid='ZRYgSHc27EqQz39ifudX', n=3, dtype='int', itype='bionty.Gene', is_type=False, hash='QW2rHuIo5-eGNZbRxHMDCw', minimal_set=True, ordered_set=False, maximal_set=False, created_by_id=1, run_id=1, space_id=1, created_at=<django.db.models.expressions.DatabaseDefault object at 0x7f22a0d59670>)
• parsing feature names of slot 'obs'
✓ 1 unique term (100.00%) is validated for name
→ returning existing schema with same hash: Schema(uid='wXSgcgUlZx76Nz1Snb1O', name='my_obs_schema', n=1, itype='Feature', is_type=False, hash='3POoh4DkKPQbuWp9wKDkvw', minimal_set=True, ordered_set=False, maximal_set=False, created_by_id=1, run_id=1, space_id=1, created_at=2025-02-26 20:55:27 UTC)
! updated otype from None to DataFrame
✓ linked: Schema(uid='wXSgcgUlZx76Nz1Snb1O', name='my_obs_schema', n=1, itype='Feature', is_type=False, otype='DataFrame', hash='3POoh4DkKPQbuWp9wKDkvw', minimal_set=True, ordered_set=False, maximal_set=False, created_by_id=1, run_id=1, space_id=1, created_at=2025-02-26 20:55:27 UTC)
✓ saved 1 feature set for slot: 'var'
Create a collection using Collection.
collection = ln.Collection([artifact, artifact2], key="my-RNA-seq-collection").save()
collection.describe()
collection.view_lineage()
Show code cell output
Collection └── General ├── .uid = 'emCEehEd1G39jzkX0000' ├── .key = 'my-RNA-seq-collection' ├── .hash = 'UK1TJC1WQ4Fe8zLDgZ2HSw' ├── .created_by = anonymous ├── .created_at = 2025-02-26 20:55:32 └── .transform = 'Introduction'

# if it's small enough, you can load the entire collection into memory as if it was one
collection.load()
# typically, it's too big, hence, open it for streaming (if the backend allows it)
# collection.open()
# or iterate over its artifacts
collection.artifacts.all()
# or look at a DataFrame listing the artifacts
collection.artifacts.df()
Show code cell output
| uid | key | description | suffix | kind | otype | size | hash | n_files | n_observations | _hash_type | _key_is_virtual | _overwrite_versions | space_id | storage_id | schema_id | version | is_latest | run_id | created_at | created_by_id | _aux | _branch_code | |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| id | |||||||||||||||||||||||
| 3 | g5mV0h4kfKxHIsE40000 | None | my RNA-seq | .h5ad | dataset | AnnData | 19240 | 9KxC2jVWsUrREWhcS15_Sg | None | 3 | md5 | True | False | 1 | 1 | 4 | None | True | 1 | 2025-02-26 20:55:29.315000+00:00 | 1 | None | 1 |
| 4 | xToOIN8U3fzMuQuZ0000 | my_datasets/my_rnaseq2.h5ad | None | .h5ad | dataset | AnnData | 19240 | Hqpz4VFmt1KN_O9MdaK75w | None | 3 | md5 | True | False | 1 | 1 | 4 | None | True | 1 | 2025-02-26 20:55:32.077000+00:00 | 1 | None | 1 |
Directly train models on collections of AnnData.
# to train models, batch iterate through the collection as if it was one array
from torch.utils.data import DataLoader, WeightedRandomSampler
dataset = collection.mapped(obs_keys=["cell_medium"])
sampler = WeightedRandomSampler(
weights=dataset.get_label_weights("cell_medium"), num_samples=len(dataset)
)
data_loader = DataLoader(dataset, batch_size=2, sampler=sampler)
for batch in data_loader:
pass
Read this blog post for more on training models on sharded datasets.
Design¶
World model¶
Teams need to have enough freedom to initiate work independently but enough structure to easily integrate datasets later on
Batched datasets (
Artifact) from physical instruments are transformed (Transform) into useful representationsLearning needs features (
Feature,CellMarker, …) and labels (ULabel,CellLine, …)Insights connect dataset representations with experimental metadata and knowledge (ontologies)
Architecture¶
LaminDB is a distributed system like git that can be run or hosted anywhere. As infrastructure, you merely need a database (SQLite/Postgres) and a storage location (file system, S3, GCP, HuggingFace, …).
You can easily create your new local instance:
lamin init --storage ./my-data-folder
import lamindb as ln
ln.setup.init(storage="./my-data-folder")
Or you can let collaborators connect to a cloud-hosted instance:
lamin connect account-handle/instance-name
import lamindb as ln
ln.connect("account-handle/instance-name")
library(laminr)
ln <- connect("account-handle/instance-name")
For learning more about how to create & host LaminDB instances on distributed infrastructure, see Install & setup. LaminDB instances work standalone but can optionally be managed by LaminHub. For an architecture diagram of LaminHub, reach out!
Database schema & API¶
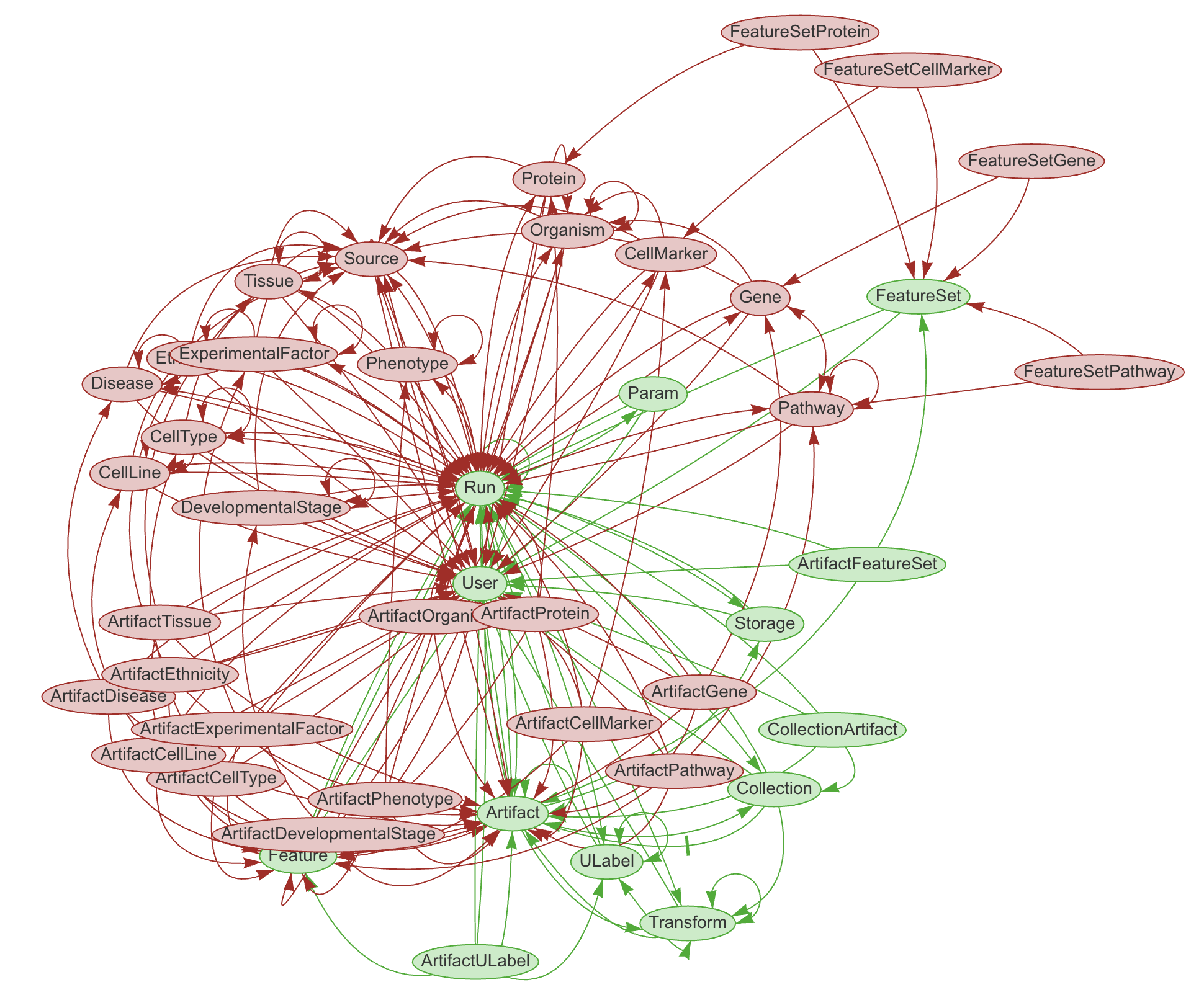
LaminDB provides a SQL schema for common metadata entities: Artifact, Collection, Transform, Feature, ULabel etc. - see the API reference or the source code.
The core metadata schema is extendable through modules (see green vs. red entities in graphic), e.g., with basic biological (Gene, Protein, CellLine, etc.) & operational entities (Biosample, Techsample, Treatment, etc.).
What is the metadata schema language?
Data models are defined in Python using the Django ORM. Django translates them to SQL tables. Django is one of the most-used & highly-starred projects on GitHub (~1M dependents, ~73k stars) and has been robustly maintained for 15 years.
On top of the metadata schema, LaminDB is a Python API that models datasets as artifacts, abstracts over storage & database access, data transformations, and (biological) ontologies.
Note that the schemas of datasets (e.g., .parquet files, .h5ad arrays, etc.) are modeled through the Feature registry and do not require migrations to be updated.
Custom registries¶
LaminDB can be extended with registry modules building on the Django ecosystem. Examples are:
bionty: Registries for basic biological entities, coupled to public ontologies.
wetlab: Registries for samples, treatments, etc.
If you’d like to create your own module:
Create a git repository with registries similar to wetlab
Create & deploy migrations via
lamin migrate createandlamin migrate deploy
Repositories¶
LaminDB and its plugins consist in open-source Python libraries & publicly hosted metadata assets:
lamindb: Core package.
bionty: Registries for basic biological entities, coupled to public ontologies.
wetlab: Registries for samples, treatments, etc.
usecases: Use cases as visible on the docs.
All immediate dependencies are available as git submodules here, for instance,
lamindb-setup: Setup & configure LaminDB.
lamin-cli: CLI for
lamindbandlamindb-setup.
For a comprehensive list of open-sourced software, browse our GitHub account.
lamin-utils: Generic utilities, e.g., a logger.
readfcs: FCS artifact reader.
nbproject: Light-weight Jupyter notebook tracker.
bionty-assets: Assets for public biological ontologies.
LaminHub is not open-sourced.
Influences¶
LaminDB was influenced by many other projects, see Influences.